How do we characterize protein dynamics? Our preferred method is heteronuclear NMR spectroscopy, which is uniquely suited to study both structure and dynamics in proteins and other biological macromolecules. A major advantage of NMR is that spectroscopic probes are distributed uniformly throughout the biomolecule, such as NH or CH atom pairs, providing large amounts of molecular information. To gain information on protein dynamics, NMR spin relaxation is highly sensitive to molecular motion over a range of timescales.
We look at the relaxation properties of 15N, 13C, 1H, and 2H spins located throughout the protein scaffold, and interpret these in terms of amplitudes and timescales of individual bond vectors. Slower motions on the microsecond-millisecond timescale can be detected to yield site-specific kinetic, thermodynamic, and structural information on the switching between discrete conformational states. In many cases, these NMR-relaxation dynamics are used to complement other structural data from X-ray crystallography, or thermodynamic and kinetic biophysical measurements using methods such as fluorescence spectroscopy, calorimetry, amide hydrogen exchange, and molecular dynamics simulations. With the larger systems now under study (CM, TS), methyl-TROSY based experiments have become indispensable.
“Slow dynamics”
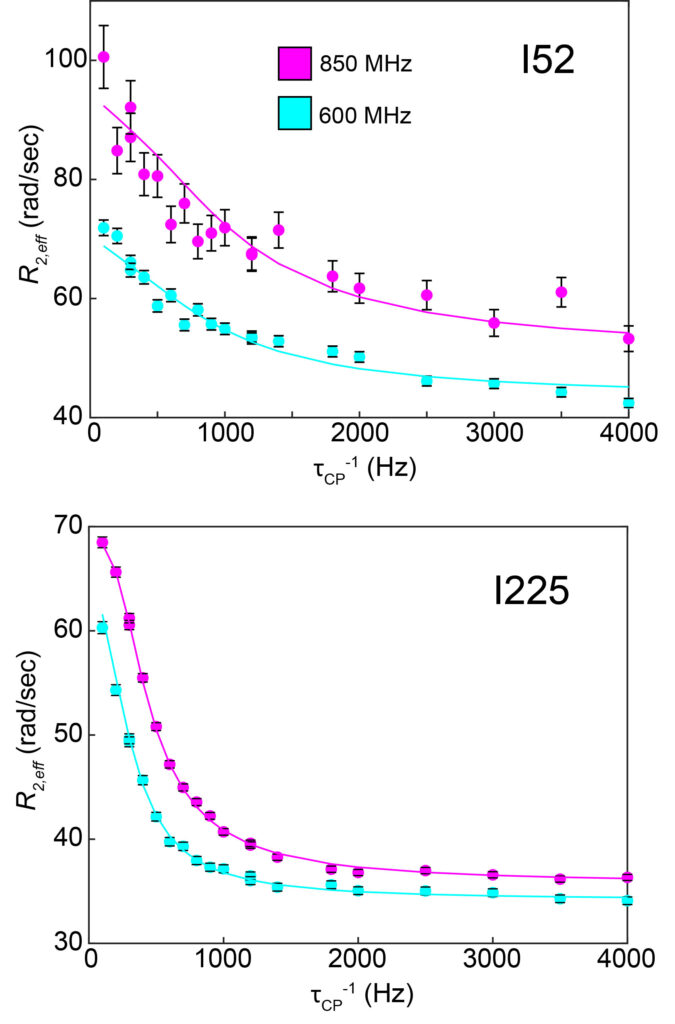
CPMG (Carr-Purcell-Meiboom-Gill) “relaxation dispersion” experiments are analyzed to characterize motions on the microsecond-millisecond timescale between two or three distinct structural states. Each NMR signal (methyl groups of I52 and I225, left) can be individually analyzed to provide a site-specific picture of the dynamics. Dispersion curves are shown at two different magnetic field strengths (850 and 600 MHz).
Even though these relaxation data were collected on the same protein sample (chorismate mutase bound to tryptophan), there are different dynamic processes occurring at residues 52 and 225, with different timescales.
“Fast dynamics”
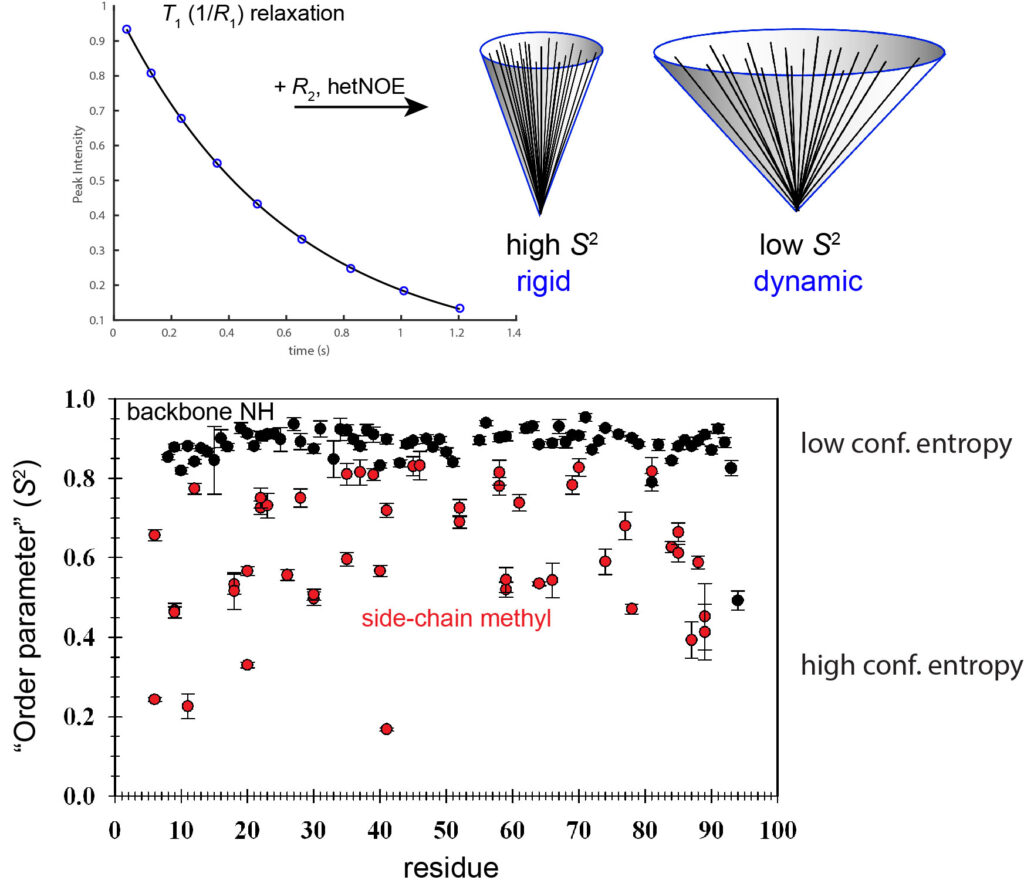
Other NMR relaxation experiments (T1, T2, etc.) can be used to capture dynamics in proteins on the picosecond-nanosecond timescale. These are typically interpreted using the Lipari-Szabo “model-free” formalism, which yield the S2 parameter (“order parameter”) for bond vector rigidity. S2 varies from 0-1, and from these measurements at multiple backbone and side-chain methyl sites, it is clear that side chains have much greater dynamics on this timescale than corresponding backbone sites. The data below were obtained on a PDZ domain from earlier Lee lab studies.